
How do I really feel about this topic? I think that I can only work out the answer to this question by writing about it.
My suspicion is that those who shout loudest about personalised medicine know least about it. I fear that the promises being made publicly are categorically not possible. My hope is that I am wrong on this.
Consider any medical data; genome, medical history, whatever. Per individual, this should be considered as a noisy sampling of the underlying data set / generator function. Now, the way to make use of this sample in a constructive manner for the individual is to aggregate the data into functional congruency sets (equivalence classes). At this point, membership of the congruency set becomes the operational level for any medical intervention / prediction. The power of this approach stems from how accurately you can map the individual’s data points to a single congruency set, and with what degree of certainty; and in the usefulness of that congruency set membership as a determinant in either classification (diagnostics) or as a lever to which treatment may be applied.
Does this functional congruency set exist? Is it typically a useful descriptor for classification? Does it correspond to suitable treatment targets?
Essentially the answer to all three questions is Yes. However this is not the endpoint of this discussion. For one thing, the more accurate answer is Yes – but only in some cases. Does it really extend to a larger space of medical issues? Or is this yet another example of the limitations of finding general solutions in a biomedical context?
From ground truth to functional classes
The first step, in a bottom-up examination of this issue is the mapping from the underlying generator function (ground truth) to the functional class. I’m going to hopscotch through this topic filling in gaps, different readers will have different requirements, let’s see how I do.
For a statistician, the actual biological measurement is considered to be noisy. There are a number of sources of noise. The two most important sources are the measurement technique (eg shotgun sequencing of the genome – yes, I know there are better methods but they all have the same statistical problems) and the availability/selection of biomarkers to read from (which is essentially technique dependent). We don’t know everything about what is going on in the underlying system – we have a sample – and we refer to the true underlying system as ground truth. Via a generator function the underlying system produces the values which we obtain in our sample.
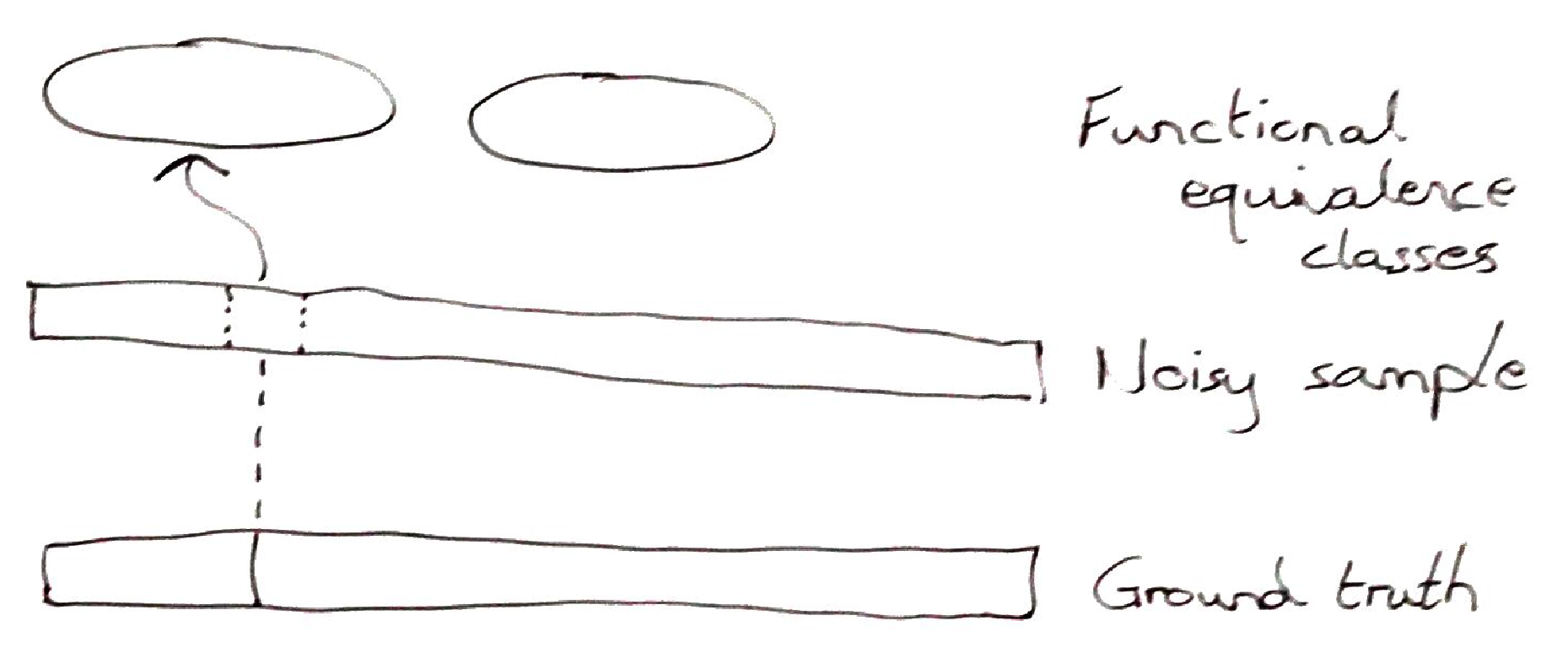
The second mapping is from the noisy sample to the functional equivalence classes. These classes are defined either by nature – via natural selection – or by our human-centric goals. I realise, while writing this, that the functional equivalence class is pretty similar to Robert Rosen‘s functional effectors in his seminal book Life Itself. The point of these equivalence classes is that they are a level of description which defines a function of the system. It can be a function inherent to normally understood biological systems, ie defined by nature. Or it can be a function which we can define in order to biochemically treat an acute or chronic illness.
Attribution (classification) of the noisy sample to the functional equivalence classes is a second statistical process. The definition of these equivalence classes is fuzzy at best. It sort of reminds me of one-sided classification, a problem I worked on for about 9 months back in 2005. We develop, over time, examples of things that we’re looking for – but we don’t know how many other sets are out there, nor how closely they map to our target set. So attribution must rely on some form of regularisation. And once we define the regularisation metric, we predefine the space of acceptable outcomes. This is fine, with one major caveat, most medical decision makers are unaware of this structural limitation and the modelling practitioners, who do understand the limitations, are rarely included in the decision making process.
From functional equivalence class to treatment
The description of how samples are assigned to functional equivalence classes, in the previous section, was a bottom-up approach. But we should also look at it from a top-down or macro-perspective, in order to understand the impacts on (personalised) treatment.
We can look at the process of assigning samples to equivalence classes from the following perspectives: (i) with what degree of certainty can we assign the sample to the class? (ii) if the sample is probabilistically assigned to multiple classes (which it should be) what are the implications of a misassignment in a treatment? This leaves one final question of particular relevance, are all functional equivalence classes clinically targetable?
The process of assigning samples to functional equivalence classes is statistical. Furthermore, we are projecting into a space which is entirely abstract and unmapped. We don’t even have any real concept of proximity in this space. We can say, based on our training set and the regularisation assumptions, that the likelihood of our new sample belonging to a particular class is p. But we do not know anything about samples which are just a little bit different from the training set. By the assumptions of regularisation we are likely to make some kind of linear mapping from our projection to the prediction classes. Since biology is structurally highly nonlinear this will work, for a while, until it breaks completely. And, unfortunately, we cannot predict when it will break.
Our best-case scenario is that there are only a small number of outcomes which are truly of interest to us. And that these are well separated in the parameter space.
In order to scale-up such a prediction system, we would need either (i) a thorough map of the functional equivalence classes, providing an understanding of when two classes are likely to have strong interactions, or (ii) a regularisation technique which ‘naturally’ accounts for the structured nature of many biological interactions.
What about the issue of classifying a sample as belonging to two or more classes? For any credible inference technique this is a strong likelihood. In this case, the issue is, what will be done after the classification?
If the prediction splits across categories which do not have strong interactions (or counter-actions) then it’s probably not a big deal. You can, maybe, treat both in parallel – or just prioritise the worst-case scenario. Here we are dealing with the shallow-end of what doctors call differential diagnosis.
The hard case is when two functional classes are predicted, but treatment on one is likely to have strong but countermanding interactions with any treatment on the other. I don’t have a particularly satisfying answer to this case. A typical response at this point, from an inference perspective, is that further data points are required – hopefully leading to a better classification. But more data may not improve the classification.
What is particularly hard in this circumstance is that you don’t know if you are in this situation because of the noisiness of the sampling and prediction hierarchy (described above) or because some biological processes are inherently intertwined and not always to the patient’s advantage.
The final consideration in this section is particular to the use of personalisation approaches in drug design. How many of the functional equivalence classes are targetable?
The choice of language – functional equivalence class – is highly deliberate. It implies that hitting any target in this class will have the same functional outcome. Obviously, I am not going into the minutiae of dosing, etc in this article – so there is a bit of hand-sweeping generalisation here. So, in theory, if you can target any aspect of the functional class for a given individual you can generate the desired health outcome.
I have already discussed, briefly, how data is mapped to these classes. But the identification of functional classes of interest is a huge, and unexplored topic. Furthermore, what is the coverage of these functional classes, in the population, when it comes to particular illnesses?

For most of these questions, we don’t have an answer right now. We gain insights through analysing the method-of-action of existing drugs. We know that many drugs only work on subsets of the population. So identifying these subsets would be a valuable first win for personalised medicine. (It is also the approach being taken by efforts to personalise oncology by sequencing tumours.)
But identifying an abstract functional equivalence class for an illness for which we have no current treatment – or for which we wish to develop a new method of treatment – involves searching while being metaphorically completely in the dark. We don’t even know how to recognise what we’re looking for when we find it.
What does this all mean for Personalised Medicine?
This article has been a thought experiment for me. I have been working on the edge of personalisation in medicine for a number of years. The process of formalising my thoughts will hopefully allow me to better understand my own macro-perspective of this toolset.
My first take-home is that the entire hierarchy, which I have described above, is statistical. I have another version of the hierarchy which looks more like a tree or a systems biology diagram, but it still leads to exactly the same conclusions! Given that every level in this description is statistical I can say with a degree of comfort that personalisation, as it is currently understood, will never mean 100% individualisation. The statistical system relies too much on chunking of people, biomarkers and mechanisms into similar categories to provide a completely individualised prediction.
The practice in machine learning is to accumulate data then to try out techniques of interest and see if any provide meaningful results. With regards to current machine learning techniques being applied in this area, I see two outcomes of this article for the machine learning practitioner. Firstly, I see strong hints as to why tree-based methods are currently working better than DNNs (hint: it’s not because of the hierarchy which I constructed, but rather because of the structure of biological data likely via natural selection). And secondly, my analysis points to areas where efforts should be focused, eg. either better understanding the functional space or finding appropriate regularisation methods, if these techniques are ever to pay off in personalising medicine.
Going forward, I think that an explicit expression of the hierarchy, which I have described, might help practitioners to better understand the points in the process at which we are being explicitly statistical, and hence the degree of individualisation which it is therefore possible to expect. This wasn’t clear to me before writing this article and I doubt that it is obvious to too many others either.
In order to use precision medicine approaches in the pharmaceutical industry, I think it is vitally important that decision makers in the drug development process better understand the formal link between the equivalence class and line-of-treatment. For me, as a mathematician, I tend to rely considerably on formal thinking. This skillset and the related insight does not naturally transfer to other people. Given this, we really need to develop new techniques for visualisation of decision making which highlight inherent knowledge which might not be so obvious to all participants.
One Reply to “Personalised Medicine – A statistical theory approach”